

Biomolecular dynamics simulations are crucial for life sciences, offering insights into molecular interactions. While classical molecular dynamics (MD) simulations are efficient, they lack chemical precision. Methods like density functional theory (DFT) achieve high accuracy but are too computationally intense for large biomolecules. MD simulations allow observation of molecular behavior, with classical MD using interatomic potentials and ab initio MD (AIMD) deriving forces from electronic structures. AIMD’s scalability issues limit its use in biomolecular studies. Machine learning force fields (MLFFs), trained on DFT-level data, promise accuracy at lower costs, though generalization across varied molecular conformations remains challenging.
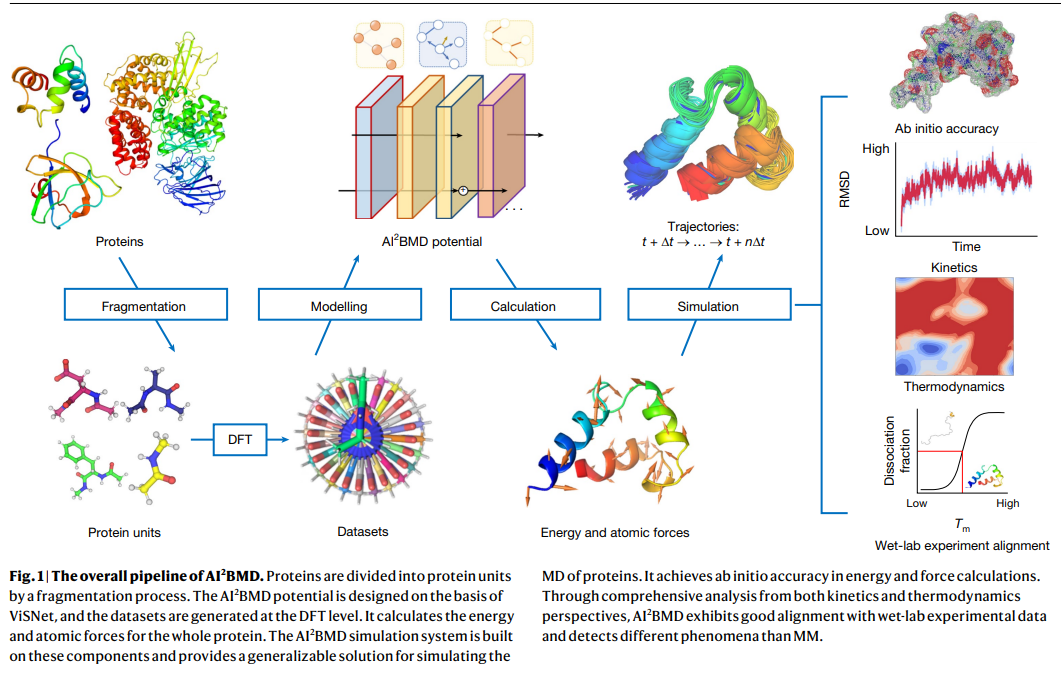
Researchers from Microsoft Research in Beijing introduced AI2BMD, an AI-based system for simulating large biomolecules with ab initio accuracy. AI2BMD utilizes a protein fragmentation technique and a machine learning force field, allowing it to accurately compute energy and forces for proteins with over 10,000 atoms. This system is vastly more efficient than traditional DFT, reducing simulation times by orders of magnitude. AI2BMD can conduct hundreds of nanoseconds of simulations, capturing protein folding, unfolding, and conformational dynamics. Its thermodynamic predictions align closely with experimental data, making it a valuable tool for complementing wet lab experiments and advancing biomedical research.
The protein fragmentation approach builds on the foundational structure of amino acids in proteins, where each amino acid contains a main chain of atoms (Cα, C, O, N, and H) and a distinct side chain. To create a model that applies broadly to various proteins, each amino acid is treated as a dipeptide, capped with Ace and Nme groups at its ends. This approach, based on overlapping fragments of dipeptides, helps ensure comprehensive protein coverage. Using a sliding window, protein chains are divided into these dipeptides, where each fragment includes main chain atoms and partial atoms from adjacent amino acids. This method accurately calculates protein energies and atomic forces by adding hydrogens as required for Cα bonds and optimizing positions using a quasi-Newton algorithm. This generalizable method allows the systematic application to all proteins, reducing complexities while maximizing model accuracy.
The training dataset for the AI2BMD potential involves sampling millions of dipeptide conformations to capture the variety in protein structures. A deep learning model called ViSNet was trained using this extensive dataset to predict the energy and atomic forces based on atomic numbers and coordinates. The model used specific hyperparameters to optimize accuracy and was trained with early-stopping techniques. Simulations based on the AI2BMD potential are processed using a cloud-compatible AI-driven simulation program, enabling flexible deployment across computing environments. This system supports parallelized simulation processes and automatically preserves progress on cloud storage, ensuring robust and efficient handling of protein dynamics modeling.
AI2BMD showcases significant potential in protein property estimation, especially for thermodynamic analysis of fast-folding proteins. AI2BMD could categorize structures into folded and unfolded states by simulating various protein types and accurately predicting potential energy values. Its melting temperature (Tm) estimations for proteins like the WW domain and NTL9 closely matched experimental data, frequently outperforming traditional molecular mechanics (MM) methods. Additionally, AI2BMD’s calculations for free energy (ΔG), enthalpy, and heat capacity were highly consistent with experimental findings, reinforcing its accuracy. This robustness in thermodynamic estimation highlights AI2BMD’s value as an advanced tool for protein analysis.
In addition to thermodynamics, AI2BMD proved effective in alchemical free-energy calculations, such as pKa prediction, and is valuable in biochemical research. Unlike traditional QM-MM methods that restrict calculations to preset regions, AI2BMD’s ab initio approach allows full-protein modeling without boundary inconsistencies, making it versatile for complex proteins and dynamic states. Although AI2BMD’s speed is still slower than classical MD, future optimizations and applications to other biomolecular systems could enhance its efficiency. AI2BMD’s adaptability makes it a promising tool for drug discovery, protein design, and enzyme engineering, offering highly accurate simulations for various biomolecular applications.
Check out the Paper and Details. All credit for this research goes to the researchers of this project. Also, don’t forget to follow us on Twitter and join our Telegram Channel and LinkedIn Group. If you like our work, you will love our newsletter.. Don’t Forget to join our 55k+ ML SubReddit.
[Sponsorship Opportunity with us] Promote Your Research/Product/Webinar with 1Million+ Monthly Readers and 500k+ Community Members
The post AI2BMD: A Quantum-Accurate Machine Learning Approach for Large-Scale Biomolecular Dynamics appeared first on MarkTechPost.



